An adaptive design approach for defects distribution modeling in materials from first-principle calculations
Abstract
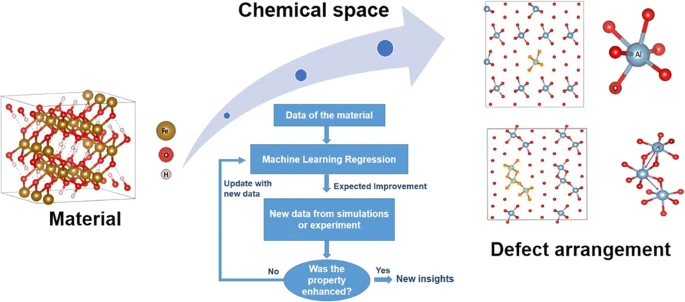
Designing and understanding the mechanism of non-stoichiometric materials with enhanced properties is challenging, both experimentally and even computationally, due to the large number of chemical spaces and their distributions through the material. In the current work, it is proposed a Machine Learning approach coupled with the Efficient Global Optimization (EGO) method—an Adaptive Design (AD)—to model local defects in materials from first-principle calculations. Our method takes into account the smallest sample set as possible, envisioning the material defect structure relationship with target properties for new insights. As an example, the AD framework allows us to study the stability and the structure of the modified goethite (Fe0.875Al0.125OOH) by considering a proper defect distribution, from first-principle calculations. The chemical space search for the modified goethite was evaluated by starting from different sizes and configurations of the samples as well as different surrogate models (ANN and Gaussian Process; GP), acquisition functions, and descriptors. Our results show that the same local solution of several defect arrangements in Fe0.875Al0.125OOH is found regardless of the initial sample and regression model. This indicates the efficiency of our search method. We also discuss the role of the descriptors in the accelerated global search for defects in material modeling. We conclude that the AD method applied in material defects is a successful approach in automating the search within huge chemical spaces from first-principle calculations by considering small samples. This method can be applied to mechanistic elucidation of non-stoichiometric materials, solid solutions, alloys, and Schottky and Frenkel defects, essential for material design and discovery.